ГЛАВА 14. ЭПИЛЕПСИЯ
ГЛАВА 14. ЭПИЛЕПСИЯ
Эпилепсия - хроническое заболевание головного мозга, проявляю- щееся повторными непровоцированными приступами с нарушением двигательных, чувствительных, вегетативных, когнитивных, психических функций, обусловленных чрезмерными нейрональными разрядами в сером веществе коры головного мозга.
Представленное определение содержит два важных положения: 1) только повторные приступы являются основанием для установления диагноза эпилепсии; 2) к эпилепсии относятся спонтанные, непрово- цируемые приступы (исключение составляют рефлекторные формы, например, фотосенситивная эпилепсия). Не являются эпилепсией фебрильные судороги, а также судороги, возникающие при острых заболеваниях головного мозга (например, при энцефалите, субдуральной гематоме, остром нарушении мозгового кровообращения и пр.).
Современные представления о заболевании начали складываться только с конца XIX в. Дж. Джэксон в 1888 г. определял эпилепсию как «...случайное, внезапное и чрезмерное локальное нарушение серого вещества головного мозга»; описал «ункусные атаки» (обонятельные галлюцинации при височной эпилепсии) и «сновидные состояния» (приступы с нарушением психических функций). А.Я. Кожевников (1898) разделил все формы эпилепсии на «органические» (по современной терминологии - симптоматические) и конституциональные (идиопатические). Первую попытку классификации эпилептических приступов предпринял английский невролог В. Говерс в 1903 г. Синдромологический подход в диагностике эпилепсии установили В. Леннокс в 1961 г., Х. Гасто в 1966 г. и Г. Доозе в 1980 г. Весомый вклад в изучение эпилепсии внесли отечественные ученые П.М. Сараджишвили и В.А. Карлов.
В конце XX в. эпилепсия стала излечимым заболеванием. Современная классификация эпилептических синдромов 1989 г. констатирует, что существует множество форм эпилепсии (синдромов), имеющих свои закономерности течения и прогноз развития в зависимости от того, какие электрические разряды происходят в коре головного мозга, где они локализуются, как распространяются и трансформируются и какие приступы при этом возникают у больного. В изучении эпилепсии важную роль играют методы нейровизуализации (КТ, МРТ с высоким разрешением, ПЭТ, SPECT), цифровая ЭЭГ и видео-ЭЭГ-мониторинг. В настоящее время примерно 65% случаев эпилепсии полностью излечимы; в 20% случаев это достигается хирургическими методами.
Изменилось и отношение к больным, улучшилась их социальная адаптация. Однако до сих пор многие механизмы патогенеза этого тяжелого заболевания не изучены; существует большое количество атипичных форм, значительно затрудняющих точную диагностику; по-прежнему остаются некурабельными некоторые резистентные формы эпилепсии.
Распространенность эпилепсии в общей популяции достигает 0,5-0,75%, а в детской - 1%. У 75% пациентов эпилепсия дебютирует в детском и подростковом возрасте, являясь одним из самых частых патологических состояний детской неврологии.
Все формы эпилепсии по этиологии подразделяются на идиопатические, симптоматические и криптогенные.
Для идиопатических форм характерны нормальный интеллект, отсутствие очаговых симптомов и структурных изменений головного мозга у пациента, а также наследственная предрасположенность (случаи эпилепсии у родственников). Этиология обусловлена главным образом каналопатиями - генетически детерминированной диффузной нестабильностью мембран нейронов. Идентифицированы гены трех основных моногенно наследуемых форм эпилепсии: аутосомно-доминантной лобной эпилепсии с ночными пароксизмами (локусы 20ql3.2 и 15q24), доброкачественных семейных судорог новорожденных (локусы 20ql3.2 и 8q24) и генерализованной эпилепсии с фебрильными судорогами плюс (локус 19ql3.1, мутация гена SCN1B; 2q21-q33, мутация гена SCN1A). Другие формы детерминированы несколькими генами (полигенное наследование). К ним относятся юношеская миоклоническая эпилепсия, роландическая эпилепсия, доброкачественная парциальная (семейная) эпилепсия младенчества и др. С практической точки зрения необходимо помнить, что если один из родителей болен идиопатической эпилепсией, вероятность рождения больного ребенка составит не более 10%.
Симптоматические формы эпилепсии характеризуются обязательным наличием морфологического субстрата: опухолей, кист, глиальных рубцов, аномалий мозга и аневризм. Их выявляют с помощью методов нейровизуализации.
Термин «криптогенный» («предположительно симптоматического генеза») определяет те формы эпилепсии, причина которых остается невыясненной даже при применении всех современных методов обсле- дования. Например, в случае сочетания эпилепсии с гемипарезом или врожденной умственной отсталостью предполагается симптоматический характер заболевания, но при КТили МР-исследовании изменения в мозге не выявляются.
Фокальные приступы и формы эпилепсии объясняет концепция коркового «эпилептогенного очага», играющего роль «водителя ритма». Возникший в нем гиперсинхронный разряд вовлекает большое количество нейронов коры, распространяясь на соседние участки головного мозга.
При генерализованных формах эпилепсии приступы генерализованы с самого начала, что подтверждается данными ЭЭГ (билатерально синхронное распространение на оба полушария). Патогенез генерализованных форм эпилепсии до настоящего времени недостаточно ясен. Ведущая таламо-кортикальная гипотеза объясняет возникновение первичной генерализации интегративной системой, состоящей из коры головного мозга и таламуса (таламо-кортикальный и кортико-таламический пути). Источник разрядов предположительно находится в коре головного мозга, таламо-кортикальные связи осуществляют синхронизацию генерализованных пик-волновых разрядов, а ретикулярная формация ствола (прежде всего среднего мозга) модулирует уровень «гиперчувствительности» коры к разрядам. В распространении и генерализации эпилептического разряда также принимают участие поясная извилина, орбито-фронтальная кора, амигдало-гиппокампальный комплекс, черная субстанция. При раздражении таламо-кортикальной системы на ЭЭГ может возникать генерализованная пик-волновая активность, а также билатерально синхронные пароксизмальные разряды ритмических дельта-волн.
Первично генерализованная эпилепсия возникает при условии аномально высокой возбудимости таламо-кортикальной системы. Уровень возбудимости, вероятно, детерминируется генетически и обусловлен нестабильностью мембран нейронов и невозможностью поддержания нормального градиента ионов Na, K и Cl.
Классификация эпилептических приступов была принята Международной лигой по борьбе с эпилепсией в 1981 г. в Киото (Япония). Эпилептические приступы подразделяют на: 1) фокальные (очаговые, фокальные, локальные, локально обусловленные); 2) генерализованные; 3) не классифицируемые (табл. 20).
Фокальные (фокальные, очаговые) приступы диагностируются в том случае, когда в начале пароксизма имеются четкие клинические и электрофизиологические критерии вовлечения определенных структур головного мозга. Например, при клонических судорогах половины лица и руки с одной стороны (фациобрахиальные приступы) эпилептический очаг находится в средненижних отделах передней
центральной извилины; при обонятельных галлюцинациях - в области крючка височной извилины; при фотопсиях - в коре затылочной доли; при «провалах мыслей» (дисмнестических приступах) - в лобной доле и т.д. При простых парциальных приступах сознание не нарушено. На ЭЭГ во время приступа отмечается локальный эпилептический разряд, начинающийся в соответствующей области коры большого мозга.
Очаговый приступ со вторичной генерализацией может начинаться как парциальный, но затем переходит в генерализованный, вовлекая все мышцы туловища и конечностей, с распространением эпилептиформной активности на ЭЭГ на оба полушария.
Сложные фокальные приступы протекают с нарушением сознания (во время приступа пациент не реагирует на обращенную речь, не выполняет команды, амнезирует приступ). ЭЭГ во время сложного парциального приступа выявляет одноили двусторонний эпилептический разряд, чаще в височных или лобных отведениях (табл. 21).
К генерализованным приступам относят типичные и атипичные абсансы, клонические, тонические, клонико-тонические и атонические приступы, а также миоклонии.
Таблица 20. Международная классификация эпилептических приступов (Киото, 1981)


Установлено, что эпилепсия не является единым заболеванием с различными приступами, а подразделяется на отдельные формы -
эпилептические синдромы. Они характеризуются устойчивой взаимосвязью клинических, электрических и анатомических критериев; различаются по реакции на антиэпилептическую терапию и по прогнозу (табл. 21).
Таблица 21. Изменения на ЭЭГ при разных приступах
Абсансы
Генерализованная билатерально синхронная и симметричная пик-волновая активность с частотой 3 Гц
Атипичные абсансы
Нерегулярная медленная пик-волновая активность менее 2 Гц, генерализованная, обычно асимметричная
Миоклонический приступ
Генерализованная билатерально синхронная пикили полипик-волновая активность
Тонико-клонический приступы
Генерализованная быстрая активность в виде множественных пиков и комплексов пик-волна
Межприступный период
Эпилептиформная активность может отсут- ствовать
Таблица 22. Международная классификация эпилепсий, эпилептических синдромов (Нью-Дели, 1989)
1. Локализационно-обусловленные формы эпилепсии (фокальные, локальные, фокальные)
1.1. Идиопатические (с возрастзависимым началом)
- Доброкачественная эпилепсия детского возраста с центральновисочными пиками (роландическая).
- Эпилепсия детского возраста с затылочными пароксизмами.
- Первичная эпилепсия чтения.
1.2. Симптоматические
- Хроническая прогрессирующая парциальная эпилепсия (синдром Кожевникова).
- Приступы, характеризующиеся специфическими способами провокации.
- Другие формы эпилепсии с известной этиологией или органическими изменениями в мозге.
1.3. Криптогенные



Следует отметить, что за прошедшее после 1989 г. время стало очевидно несовершенство классификации, поскольку в нее не вошли некоторые формы (например, синдром псевдоленнокса). Кроме того, многие симптоматические формы синдрома Веста и синдрома Леннокса-Гасто не относятся к генерализованной эпилепсии, поскольку представляют собой парциальную эпилепсию с феноменом вторичной билатеральной синхронизации. В 2001 г. Международная комиссия по классификации и терминологии выпустила проект новой классификации эпилептических приступов и эпилептических синдромов (табл. 22). Кроме классического деления на фокальные и генерализованные приступы, в нем указано, что в отношении многих доброкачественных и самокупирующихся эпилептических синдромов термин «эпилепсия» следует заменять на «приступы». Например, не «алкогольная эпилепсия», а «приступы, связанные с отменой алкоголя» и т.д. Описано много новых форм эпилепсии как четко установленных, введены новые термины. Термин «парциальные приступы и парциальные эпилепсии» заменен на «фокальные приступы и фокальные формы эпилепсии»; «криптогенные формы» на «вероятно симптоматические формы». В определении синдромов рекомендована замена слова «судороги» на «приступы». Понятие «приступы» значительно шире понятия «судороги», и далеко не все приступы проявляются именно судорогами. Упразднено подразделение фокальных приступов на простые и сложные в зависимости от нарушения сознания, так как в большинстве случаев оценка уровня сознания остается ориентировочной. Достоинством классификации является разработка концепции детских эпилептических энцефалопатий.
Диагностика эпилепсии включает следующий алгоритм:
1. Описание пароксизмального события (возможно исключительно по данным анамнеза).
2. Классификация приступов (анамнез, клиника, ЭЭГ, видеоЭЭГ-мониторинг).
3. Диагностика формы (анамнез, клиника, ЭЭГ, видео-ЭЭГмониторинг, нейровизуализация).
4. Установление этиологии (МРТ, кариотипирование, биохимические исследования, биопсия мышц и пр.).
5. Диагностика сопутствующих заболеваний и установление степени инвалидизации.
Диагноз эпилепсии является клинико-электро-анатомическим. В XXI в. для установления точного диагноза эпилепсии недостаточно иметь описание приступов, представленное родственниками. Необходимо электроэнцефалографическое подтверждение (электрический критерий), а также проведение методов нейровизуализации (анатомический критерий). Для точного определения диагноза и назначения правильной терапии, кроме рутинных методик, необходимо проведение длительного ЭЭГ-видеомониторинга, ночного ЭЭГ- мониторинга, высокоразрешающей МРТ в режиме 3D-визуализации и т.д.
14.1. Идиопатические фокальные формы
Доброкачественная парциальная эпилепсия детского возраста с центрально-височными пиками (роландическая эпилепсия) [РЭ] - характеризуетсяся короткими фарингооральными и гемифациальными моторными приступами, возникающими обычно при пробуждении и засыпании, а также типичными изменениями на ЭЭГ (рис. 14.1). РЭ - самая частая форма эпилепсии в детском возрасте. Показатель заболеваемости составляет 21 на 100 000 детского населения.
Заболевание начинается в возрасте от 2 до 14 лет (максимум в 7-9 лет), чаще болеют мальчики. Характерны простые фокальные приступы, возникающие в 80% случаев при пробуждении или засыпании. Приступ начинается с соматосенсорной ауры: ощущения покалывания, онемения с одной стороны в области глотки, языка, десны. Затем пациенты издают своеобразные горловые звуки типа «бульканья», «хрюканья», «полоскания горла»; отмечается гиперса- ливация и анартрия (фарингооральные приступы). Характерны судороги мимических мышц: односторонние тонические, клонические
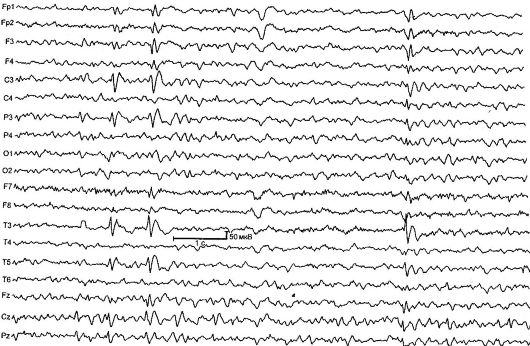
Рис. 14.1. ЭЭГ ребенка 4 лет с роландической эпилепсией
или тонико-клонические судороги мышц лица, губы, а также языка, глотки, гортани (гемифациальные приступы). У 20% больных судороги распространяются с мышц лица на гомолатеральную руку (бра- хиофациальные приступы); примерно в 8% случаев они появляются и в ноге (унилатеральные приступы). По мере развития заболевания приступы могут изменять сторонность.
Вторично-генерализованные судорожные приступы отмечаются у 25% детей. Приступы при РЭ продолжаются от нескольких секунд до 1-2 мин. Частота их в среднем - 2-6 раз в год. С течением времени они возникают все реже (даже без лечения), и у взрослых не наблюдаются.
Изменения на ЭЭГ в межприступном периоде определяются в 90% случаев, типичный паттерн - комплекс острая-медленная волна. Начальный компонент обычно состоит из трехфазной острой волны с последующей медленной волной, что создает сходство с комплексами QRST на ЭКГ. Эта активность локализуется в центрально-височных отведениях и называется «роландической» или имеет общее название - «доброкачественные эпилептиформные нарушения детского возраста» (ДЭНД). Для подтверждения диагноза РЭ важно проводить
ЭЭГ во время сна - ночной ЭЭГ-мониторинг, так как примерно у 30% детей с РЭ роландические комплексы выявляются исключительно во время сна.
Терапия. Учитывая доброкачественное течение, можно не назна- чать антиэпилептическую терапию. Однако не исключена диагностическая ошибка, а также возможность трансформации РЭ в синдром псевдоленнокса примерно в 5% случаев у детей до 7 лет. Рекомендуется начинать терапию при повторных приступах. Лечение всегда проводят одним препаратом (политерапия недопустима), начиная с производных вальпроевой кислоты (депакин, конвулекс, конвульсофин). Вальпроаты назначают с постепенным наращиванием дозы до 15- 30 мг/кг в сутки (в среднем 600-1500 мг/сут) в 2 приема.
При неэффективности или непереносимости вальпроатов назначают топирамат (топамакс) в дозе 50-150 мг/сут (3-5 мг/кг). Также применяются препараты из группы карбамазепина (тегретол, финлепсин) в средней суточной дозе 15-20 мг/кг (300-600 мг/сут). В отдельных случаях карбамазепин может привести к увеличению индекса ДЭНД на ЭЭГ и учащению приступов - феномену аггравации. В связи с этим не рекомендуется назначать карбамазепин как стартовую терапию, а также во всех случаях у детей до 7 лет. Применение барбитуратов и гидантоинов противопоказано!
Необходим контроль ЭЭГ, в том числе ЭЭГ-мониторинг сна. Ремиссия при РЭ достигается в 100% случаев к 16 годам.
Идиопатическая парциальная эпилепсия с затылочными пароксизмами (доброкачественная затылочная эпилепсия, ДЗЭ) - характеризуется приступами с нарушением зрительных функций, мигренеподобными симптомами и наличием на ЭЭГ паттерна ДЭНД в затылочной области. ДЗЭ составляет около 20% всех идиопатических парциальных форм эпилепсии детского возраста. Выделено два варианта ДЗЭ: с ранней и поздней манифестацией заболевания.
Доброкачественная затылочная эпилепсия с ранним дебютом (синдром Панайотопулоса) начинается между 1 и 13 годами, с пиком манифестации в 3-6 лет. Заболевание проявляется редкими тяжелыми приступами с вегетативными нарушениями, длительной утратой сознания и тенденцией к статусному течению. Приступы возникают во сне, особенно перед пробуждением; начинаются с рвоты, головной боли, побледнения лица, с последующим поворотом головы и глаз в сторону. Приступы обычно заканчиваются гемиконвульсивными или генерализованными судорогами. Возникают «иктальные синкопы», проявляющиеся длительной
утратой сознания и резким падением мышечного тонуса, продолжительностью от 30 мин до 7 ч, в среднем 2 ч. Большинство пациентов попадают в реанимационное отделение. «Иктальные синкопы» могут как предшествовать вторично-генерализованным тонико-клоническим судорогам, так и возникать изолированно от них. Несмотря на тяжелое статусное течение, частота подобных приступов невелика. В некоторых случаях отмечается лишь один приступ за весь период заболевания. Прогноз - абсолютно благоприятный.
Доброкачественная затылочная эпилепсия с поздним дебютом (форма Гасто) дебютирует с 3 до 15 лет, в среднем в 8 лет. Характерны простые фокальные сенсорные приступы со зрительными нарушениями в виде простых зрительных галлюцинаций (маленьких разноцветных круговых фигур), которые часто возникают в периферическом поле зрения и двигаются в противоположную очагу сторону. Приступы длятся от нескольких секунд до 1-3 мин. Галлюцинации могут возникать в одноименных половинах полей зрения. Часто отмечается версивный компонент - поворот глаз и головы контралатерально очагу при сохранном сознании. Приступы могут заканчиваться унилатеральными или вторично-генерализованными тонико-клоническими судорогами. У половины пациентов после приступа появляется интенсивная пульсирующая мигренеподобная головная боль, сопровождающаяся тошнотой и рвотой. Частота приступов обычно невелика, хотя в отдельных случаях они могут быть еженедельными. На ЭЭГ выявляются высокоамплитудные комплексы острая-медленная волна, возникающие у 2/3 пациентов только в затылочных отведениях. Морфология комплексов сходна с доброкачественными эпилептиформными нарушениями детского возраста. У 1/3 больных эпилептиформная активность может регистрироваться и в других областях (чаще в центрально-височных отведениях).
Терапия. Препаратами первого выбора в лечении ДЗЭ являются соли вальпроевой кислоты (депакин, конвулекс, конвульсофин) в средней суточной дозе 30-40 мг/кг. Препарат назначают в два приема с максимальной дозировкой в вечернее время.
При недостаточной эффективности возможна монотерапия препаратами карбамазепина (финлепсин, тегретол) в средней дозе 15- 20 мг/кг/сут или топираматом в дозе 75-200 мг/сут (3-6 мг/кг/сут).
При синдроме Панайотопулоса полная ремиссия приступов к 9 годам наступает у 92% пациентов. У больных с формой Гасто ремис- сия наблюдается в 82% случаев к 15 годам и в 100% - к 18.
Аутосомно-доминантная лобная эпилепсия с ночными приступами
является идиопатической формой. Идентифицировано 2 генных локуса, ответственных за развитие данного заболевания: 20q13.2 и 15q, но встречаются и спорадические случаи. Возраст дебюта варьирует от 2 мес до 52 лет, с максимумом на первом десятилетии жизни. Приступы у 70% пациентов начинаются с неспецифической ауры: «ознобоподобного дрожания», головной боли, слуховых галлюцинаций, головокру- жения, соматосенсорных ощущений (зуда в области туловища), после которой типичны приступы с гипермоторными автоматизмами. Они начинаются с судорожного дыхания, хрюканья, сильного крика по типу завываний. Глаза широко открыты, на лице выражение ужаса. Пациент поднимает голову, садится в кровати; появляются гипермоторные и дистонические феномены. Иногда пациент (чаще взрослый) соверша- ет хаотичные движения руками (по типу боксирующих движений) и ногами (типа педалирования); встает на четвереньки и совершает раскачивающиеся движения тазом. Сознание во время приступов обычно не нарушено. Характерно возникновение приступов исключительно во сне, они могут повторяться многократно в течение ночи в виде серии, затем происходит перерыв на несколько дней или недель и опять возобновляется серия. Продолжительность приступов - от нескольких секунд до 1 мин. В редких случаях возможно появление вторичногенерализованных пароксизмов.
ЭЭГ бодрствования неспецифична. Диагностически значимы данные ЭЭГ-мониторинга ночного сна и видео-ЭЭГ-мониторинга, которые выявляют низкоамплитудную эпилептиформную активность в виде комплекса острая-медленная волна, возникающую регионально в одном из лобных, лобно-височных отведений или бифронтально асинхронно.
Стартовое лечение начинается с препаратов карбамазепина, двукратно с максимумом перед ночным сном. Суточная дозировка - 600-1000 мг/сут (15-30 мг/кг/сут). При неэффективности назначается топирамат в дозировке 100-400 мг/сут (3-10 мг/кг/сут), двукратно с максимумом перед ночным сном. Следующий этап лечения - монотерапия вальпроатами. Назначается конвулекс двукратно в дозе
900-1800 мг/сут (20-40 мг/кг/сут).
В редких случаях резистентности может быть применена политерапия, состоящая из комбинации двух базовых АЭП (вальпроевой кислоты с карбамазепином или топираматом). Медикаментозная ремиссия достигается в большинстве случаев.
14.2. Симптоматические фокальные формы эпилепсии
Симптоматическая лобная эпилепсия (СЛЭ) - локально обусловленная форма с верифицированными морфологическими нарушениями в пределах лобных долей большого мозга. Составляет 30-40% среди всех симптоматических фокальных форм эпилепсий и занимает 2-е место по частоте после височной эпилепсии (в детском возрасте может опережать височную эпилепсию по частоте встречаемости).
Этиология включает черепно-мозговые травмы, опухоли и кисты лобной доли, фокальные кортикальные дисплазии, глиоз как след- ствие перинатальной энцефалопатии, сосудистые аномалии.
В рамках СЛЭ выделяют несколько формы.
Моторная (премоторная, джексоновская) СЛЭ возникает при раздражении передней центральной извилины. Характерны простые фокальные моторные приступы с судорогами в контралатеральных очагу конечностях. «Джексоновский» марш начинается судорогами кисти или стопы, с постепенным вовлечением руки, ноги и мышц лица одноименной стороны. Нередко приступ заканчивается преходящим парезом Тодда.
Оперкулярная СЛЭ возникает при раздражении оперкулярной зоны лобной доли. Характеризуется сложными фокальными (диалептическими) приступами с оро-алиментарными автоматизмами; возможны ипсилатеральное подергивание лицевой мускулатуры, вегетативные феномены.
Орбитофронтальная СЛЭ возникает при раздражении орбитальной коры нижней лобной извилины. Характеризуется сложными фокальными, вегетативно-висцеральными приступами, пароксизмами с насильственной вокализацией, атипичными абсансами.
Дорсолатеральная (префронтальная) СЛЭ возникает из задних отделов верхней и нижней лобной извилины. Проявляется тоническими адверсивными приступами с поворотом глаз и головы в сторону, противоположную очагу; возможно также отведение и приподнима- ние руки, на которую устремлен взор больного. Нередко появление моторной афазии при локализации очага в доминантной гемисфере.
Фронтополярная СЛЭ возникает при локализации эпилептогенного очага в области полюса лобных долей. Представлена простыми парциальными приступами с нарушением когнитивных функций (наплыв мыслей, «провал» мыслей, изменение течения времени) и сложными парциальными (диалептическими) приступами.
Цингулярная СЛЭ наблюдается при раздражении передней части поясной извилины. Проявляется сложными парциальными приступами с жестовыми автоматизмами, ипсилатеральными моргательными движениями, а также «лимбическими пароксизмами»: выражением страха, покраснением лица, нарушением эмоциональной сферы - дисфорией.
СЛЭ, исходящая из дополнительной моторной зоны (премоторная СЛЭ), - одна из наиболее частых форм лобной эпилепсии, характеризуется короткими постуральными асимметричными тоническими приступами (спазмами), появляющимися билатерально в проксимальных отделах конечностей (например, типа «позы фехтовальщика»). Приступы преимущественно ночные, возникают серийно. Также наблюдаются приступы с остановкой речи при ясном сознании или вокализацией в виде криков, завывающих звуков. Возможны приступы со стереотипными гипермоторными автоматизмами: хаотичные движения руками (по типу боксирования), ногами (педалирующие движения), тазом.
Приступы короткие, с непродолжительным или неполным выключением сознания, минимальной постиктальной спутанностью, серий- ным циклолептическим течением и преимущественным возникновением в ночное время.
Результаты неврологического обследования зависят от этиологии СЛЭ. При обширном поражении лобной доли (например, объемном образовании) выявляется гемипарез на стороне, противоположной очагу (высокие рефлексы, патологические рефлексы); возможна гемиатаксия. Нередко формируется нарушение поведения по типу «лобной психики».
ЭЭГ в межприступном периоде малоинформативно или неспецифично. Предпочтительнее длительный ЭЭГ-мониторинг (и обязательно во время сна), который выявляет региональные эпилептиформные паттерны (острая-медленная волна), продолженное региональное замедление в одном из лобных отведений, феномен вторичной билатеральной синхронизации.
Для выявления структурного дефекта проводят МРТ.
Стартовое лечение начинается с топирамата (топамакса) в начальной дозе 12,5-25 мг/сут. Дозу постепенно увеличивают на 12,5-25 мг 1 раз в неделю до 50-500 мг/сут (3-10 мг/кг/сут), в 2 приема (утром и вечером) с интервалом в 12 ч. Препарат второго выбора - карбамазепин, применяют в дозе 600-1800 мг/сут (15-35 мг/кг/сут), 2 раза в сутки. Карбамазепин и окскарбазепин особенно эффективны при диалептических приступах. При «псевдогенерализованных присту-
пах» и феномене вторичной билатеральной синхронизации на ЭЭГ карбамазепин противопоказан, поскольку способен аггравировать приступы.
Средства третьего выбора - препараты вальпроевой кислоты (конвулекс, депакин, конвульсофин) применяют в дозе 1000-3000 мг/сут (30-60 мг/кг/сут), 2 раза в сутки.
При неэффективности трех базовых препаратов рекомендована политерапия - комбинация топирамата или вальпроатов с сукци- нимидами. Этосуксимид (суксилеп) назначают в дозировках 500- 1000 мг/сут (20-40 мг/кг/сут) в 3 приема. В остальных случаях назначают комбинацию базовых АЭП: топирамат + вальпроаты, вальпроаты + карбамазепин, карбамазепин + топирамат.
Резервные препараты при политерапии - ламотриджин (ламиктал) и леветирацетам (кеппра). Ламотриджин (3-7 мг/кг/сут) применяют только в комбинации с базовыми АЭП. Средние дозировки - 100-400 мг/сут в комбинации с топираматом или карбамазепином и 100-200 мг/сут с вальпроатами. Леветирацетам эффективен в комбинации с базовыми АЭП в дозе 1000-4000 мг/сут (30-60 мг/ кг/сут) при фокальных моторных и вторично-генерализованных приступах.
Прогноз заболевания при СЛЭ всегда серьезный, что связано с наличием структурного дефекта в коре, гемипареза и выраженных когнитивных нарушений. Медикаментозная ремиссия достигается только у 20% больных. В остальных случаях удается существенно снизить частоту приступов. При резистентных приступах применяется хирургическое лечение. Основной вид оперативного вмешательства - фокальная кортикальная резекция.
Симптоматическая височная эпилепсия (СВЭ) - локально обусловленная форма с известной этиологией и морфологическими нарушениями в височных долях головного мозга (склероз аммонова рога, доброкачественные врожденные опухоли височной доли, фокальные корковые дисплазии, последствие перинатального поражения). Выделяют две основные формы СВЭ: лимбическую (синонимы: палеокортикальная, амигдало-гиппокампальная) и неокортикальную (синоним: латеральная).
В 75% случаев приступы начинаются с ауры. Следует четко определить понятие ауры и отграничить ее от предвестников эпи- лептического приступа. Под аурой (от греч. - дуновение) следует понимать клинические феномены, которые возникают сами по себе
или перед вторично-генерализованным или парциальным приступом. Аура обусловлена локальным эпилептическим разрядом в определенном участке коры большого мозга и по сути является простым парциальным приступом. Характер ауры указывает на локализацию очага. Выделяют следующие виды ауры: соматосенсорную, зрительную, обонятельную, вкусовую, слуховую, головокружение, психическую, вегетативную, брюшную (абдоминальную). Предвестники возникают за многие минуты, часы или дни до эпилептического приступа, про- являются обычно психическими или вегетативными симптомами, не сопровождающимися локальными кортикальными разрядами.
Амигдало-гиппокампальная (палеокортикальная, лимбическая) - наиболее частая форма, составляет около 65% среди всех случаев СВЭ. В основе заболевания чаще лежит склероз (глиоз) медиобазальных отделов височной доли вследствие перинатального поражения или атипичных фебрильных судорог. Заболевание обычно начинается с длительных, нередко гемиклонических, фебрильных судорог в возрасте до 3 лет. Далее следует период мнимого благополучия - при- ступы отсутствуют вплоть до препубертатного периода. Наиболее типичны (70% случаев) сложные фокальные приступы с выключением сознания (диалептические) или автоматизмами (аутомоторные). При диалептических приступах больной внезапно прекращает двигательную активность, застывает с широко раскрытыми глазами, взгляд выражает изумление или испуг («staring gaze»).
Для СВЭ характерны автоматизмы в виде жестов (потирание рук, пальцев, сжимание кисти, перебирание одежды) и оро-алиментарных действий (причмокивание, сглатывание, облизывание). Автоматизмы в кисти наблюдаются на стороне очага, а дистоническая установка пальцев кисти - на противоположной. Продолжительность аутомоторных приступов от 30 сек до 3 мин, они быстро учащаются и становятся резистентными к терапии.
Нередко приступы сопровождаются нарушением вегетативных функций. Особенно характерны эпигастральные пароксизмы при ясном сознании. Пациент ощущает боль, распирание, дискомфорт в области пупка; возможно отхождение газов. Это «восходящее эпилептическое ощущение» поднимается из живота вверх к горлу, сопровождается чувством сжатия шеи, после чего возможно выключение сознания.
Также характерны простые фокальные приступы с нарушением психических функций: сновидные состояния Джексона («dreamy states»), проявляющиеся внезапными своеобразными ощущениями
«снов наяву»; ощущение «уже виденного» или «никогда не виденного»; возникновение дереализации (ощущение нереальности окружающего) или деперсонализации (нарушение восприятия собственной личности). При вовлечении миндалевидного комплекса появляются короткие приступы немотивированного страха, дисфории, агрессии.
Латеральная (неокортикальная) СВЭ возникает при поражении верхнелатеральных отделов височной доли. Возможны следующие виды приступов: слуховые галлюцинации (пароксизмальные ощущения шума, музыки, голосов); зрительные галлюцинации (пароксизмальное появление сложных ярких панорамных зрительных образов, нередко с элементами воспоминания прошедших событий); приступы несистемного головокружения, часто в сочетании с вегетативными проявлениями (бледностью кожи, гипергидрозом, тахикардией); пароксизмальная сенсорная афазия при локализации эпилептогенного очага в доминантном полушарии; «височные синкопы» с выключением сознания, обмяканием и медленным падением без судорог.
При неврологическом осмотре нередко выявляются пирамидные симптомы контралатерально очагу: нарушение функции VII и XII черепных нервов, асимметрия мышечного тонуса, анизорефлексия, патологические рефлексы. У взрослых пациентов при длительном течении заболевания развиваются личностные и когнитивные нарушения, обозначаемые термином «глишроидия»: вязкость, тугоподвижность, инертность мышления, сложности переключения, «застревание» на мелочах, стойкость аффекта; снижение памяти и внимания.
ЭЭГ в межприступном периоде в 50% случаев - без патологических изменений. Пик-волновая активность в височных долях реги- стрируется не более чем у 20% пациентов.
На МРТ в коронарной проекции могут выявляться склероз гиппокампа, расширение нижнего рога бокового желудочка, уменьшение в объеме пораженной височной доли, в ряде случаев - фокальная корковая дисплазия.
Лечение начинают с препаратов карбамазепина (финлепсин ретард, тегретол СR), в дозе 600-1800 мг/сут (15-35 мг/кг/сут) в 2 приема с 12-часовым интервалом или в 3 приема с 8-часовым интервалом. Окскарбазепин (трилептал) назначают в дозе 600- 2400 мг/сут (20-40 мг/кг/сут). Препарат второго выбора - топирамат, назначают, постепенно увеличивая дозу до 100-400 мг/сут (4-8 мг/ кг/сут), 2 раза в день.
Средства третьего выбора - препараты вальпроевой кислоты применяют в дозе 1000-3000 мг/сут (30-70 мг/кг/сут) в 2 или 3 приема с равными временными интервалами.
При неэффективности трех базовых препаратов рекомендована политерапия: комбинации карбамазепина (или окскарбазепина) с вальпроатами, топираматом; вальпроатов с топираматом. Резервные препараты при политерапии - ламотриджин (3-7 мг/кг/сут, только в комбинации с базовыми АЭП) и леветирацетам.
Прогноз. Медикаментозная ремиссия достигается лишь у 1/3 больных. У остальных пациентов в большинстве случаев удается существенно снизить частоту приступов. В медикаментозно резистентных случаях применяют хирургическое лечение, в частности селективную амигдало-гиппокампотомию.
Симптоматическая затылочная эпилепсия (СЗЭ) характеризуется наличием эпилептогенного очага и морфологическими изменениями в затылочной области. Этиологическими факторами являются фокальные корковые дисплазии, последствие перинатальных поражений, окципитальные кальцификаты с целиакией, сосудистые аномалии (синдром Штурге-Вебера), MELAS, прогрессирующая миоклонус-эпилепсия с тельцами Лафоры, опухоли, ОНМК в бассейне задней мозговой артерии.
Возраст начала СЗЭ вариабелен. Констатируют следующие виды приступов: простые фокальные сенсорные со зрительными рас- стройствами (макро-, микропсии, элементарные зрительные галлюцинации), с глазодвигательными нарушениями (адверсия головы и глаз в противоположную очагу сторону, форсированное пароксизмальное моргание, нистагм); вегетативно-висцеральные (тошнота, рвота, головная боль); вторично-генерализованные судорожные. Нередко в структуре приступа (или в качестве постприступных симптомов выпадения) наблюдаются амавроз и гомонимная квадрантная гемианопсия. Характерна постприступная мигренеподобная головная боль.
При неврологическом обследовании в отдельных случаях определяются косоглазие, амблиопия, сужение полей зрения или гемианопсия. ЭЭГ-исследование в межприступном периоде у 30% больных СЗЭ не выявляет патологических изменений. Чаще определяются региональное замедление или пик-волновая эпилептиформная активность в одном из затылочных отведений или биокципитально с амплитудным преобладанием на стороне очага.
Нейровизуализация выявляет затылочные кортикальные дисплазии, локальный глиоз вследствие перенесенной перинатальной энцефалопатии (улегирия), кальцификаты, сосудистые аномалии.
Лечение начинают с препаратов карбамазепина в дозе 600- 1800 мг/сут (15-35 мг/кг/сут), в 2 приема с 12-часовым интервалом. Карбамазепин в высоких дозах особо эффективен при изолированных зрительных аурах и фокальных приступах с нарушением вегетативных функций. Многие авторы рекомендуют начинать лечение СЗЭ с окскарбазепина в дозе 600-2400 мг/сут (20-40 мг/ксут).
Препарат второго выбора - топирамат назначают в дозе 100- 400 мг/сут (5-8 мг/кг/сут) 2 раза в день. При вторичной билате- ральной синхронизации на ЭЭГ топамакс может быть стартовым препаратом.
Препарат третьего выбора - вальпроевая кислота. Средние дозировки - 1000-2000 мг/сут (30-60 мг/кг/сут), при необходимости - выше, в 2 или 3 приема.
В резистентных случаях применяется политерапия. Особенно эффективны комбинации карбамазепина (или окскарбазепина) с вальпроатами, вальпроатов с топираматом, реже - карбамазепина с топираматом. При добавлении второго препарата дозировка первого, как правило, не уменьшается. Резервные препараты при политерапии - ламотриджин и леветирацетам.
Прогноз зависит от характера структурного дефекта мозга и путей распространения возбуждения в коре. У 40-50% больных может быть достигнута стойкая медикаментозная ремиссия. В резистентных случаях СЗЭ при отсутствии эффекта от применения АЭП единственным методом реальной помощи пациентам является нейрохирургическое вмешательство - кортикальная резекция.
Эпилепсия Кожевникова и энцефалит Расмуссена (ЭК) - полиэтио- логичное заболевание, проявляющееся сочетанием миоклонических, фокальных моторных, вторично-генерализованных приступов с очаговыми неврологическими симптомами.
Заболевание впервые описал российский невролог профессор Алексей Яковлевич Кожевников под названием «epilepsia corticalis sive partialis continua». 21 января 1894 г. на заседании созданного им Московского общества неврологов и психиатров он выступил с докладом на тему «Об особом виде кортикальной эпилепсии». Доклад был основан на изучении 4 случаев кортикальной эпилепсии, наблюдаемых автором в клинике нервных болезней Москвы, и представлял собой
оригинальное описание заболевания, до того времени еще не известного. Клиническая картина болезни у всех 4 пациентов была в высшей степени схожа: «...сочетание генерализованных эпилептических при- ступов с постоянными клоническими судорогами в строго определенных частях тела. Из этих постоянных судорог развивались: 1) типичные джексоновские припадки в одной половине тела и 2) вышеупомянутые общие припадки, развивавшиеся также по джексоновскому типу». Другое название этого заболевания было предложено присутствовавшим на докладе профессором Н.Ф. Филатовым - «кожевниковская эпилепсия». В 40-е годы прошлого века была доказана взаимосвязь ЭК с весеннее-летним клещевым энцефалитом (русский энцефалит).
В 1958 г. Т. Расмуссен, Ж. Обжевски описали клинику хронического очагового энцефалита, одним из кардинальных симптомов которого была ЭК. Позже данное заболевание было названо энцефалитом Расмуссена, или синдромом Расмуссена (СР). До настоящего времени остается загадкой, при каком заболевании А.Я. Кожевников описал симптомокомплекс ЭК - при русском энцефалите или энцефалите Расмуссена. По нашему мнению, А.Я. Кожевников, практиковавший в Москве, описал свою форму эпилепсии именно при хроническом очаговом энцефалите, так как ни в одной из представленных им историй болезни нет указаний на перенесенный пациентами острый энцефалит.
Помимо клещевого энцефалита, ЭК вызывают туберкулезный менингоэнцефалит, нейросифилис, черепно-мозговая травма, опу- холи головного мозга, фокальные кортикальные дисплазии, наследственные болезни обмена.
Хронический очаговый энцефалит [энцефалит Расмуссена, синдром Расмуссена (СР)]. СР представляет собой тяжелое заболевание головного мозга - хронический прогрессирующий очаговый энцефалит. Заболевание характеризуется триадой клинических симптомокомплексов: эпилептическими приступами (по типу эпилепсии Кожевникова), двигательными нарушениями (центральный гемипарез) и расстройством высших психических функций. Этиология неизвестна, предположительно заболевание относят к медленным нейроинфекциям вирусной этиологии, но вирус не идентифицирован.
Дебют в детском возрасте - от 1 года до 14 лет, с пиком в 5-6 лет с эпилептических приступов (фокальных моторных или вторичногенерализованных, реже - диалептических); в 20% случаев - с эпилеп-
тического статуса. Нередко отмечается соматосенсорная аура (жжение, покалывание, онемение). Уже на начальных этапах заболевания развивается преходящий постиктальный монопарез (или гемипарез) - парез Тодда. Обычно через несколько месяцев после появления первых фокальных приступов к ним присоединяются длительные (до нескольких дней), а затем постоянные, локализованные в одной половине туловища и конечностей миоклонические пароксизмы, которые могут трансформироваться в генерализованные судороги. Указанный симптомокомплекс представляет собой эпилепсию Кожевникова. С течением времени эпилептический миоклонус распространяется на все конечности, лицевую мускулатуру, мышцы передней брюшной стенки и становится постоянным, не исчезая и во сне. Развивается стойкий гемипарез. Присоединяются нарушение чувствительности по проводниковому типу и выпадение полей зрения. Нарастают когнитивные нарушения, дизартрия. В 25% случаев возможно ожирение, преждев- ременное половое развитие.
На ЭЭГ в развернутой стадии заболевания в 100% случаев наблюдается прогрессирующее замедление основной активности фона, продолженное региональное замедление (в лобно-височных отведениях); продолженная пик-волновая активность. По мере прогрессирования эпилептиформная активность возникает диффузно.
Нейровизуализация имеет решающее значение в диагностике. При МРТ головного мозга в динамике отмечается нарастание гемиа- трофии. Атрофия обычно начинается с теменно-височной области в виде локального расширения сильвиевой щели и с течением времени распространяется «подобно масляному пятну по листу пергаментной бумаги», захватывая «здоровое» полушарие.
ЭК относится к резистентным эпилептическим синдромам. Стартовая терапия - вальпроаты (депакин, конвулекс, конвульсофин) в высоких дозах: до 50-100 мг/кг/сут. Далее рекомендуется комбинация вальпроатов с леветирацетамом или топираматом. Показана эффективность леветирацетама при фокальных моторных, вторично-генерализованных и миоклонических приступах в рамках ЭК, его дозировка - 30-70 мг/кг/сут. Дозировка топирамата составляет около 10 мг/кг/сут. В развернутой стадии заболевания возможно применение барбитуратов (фенобарбитал 5-8 мг/кг/сут). Добавление этосуксимида (до 30 мг/кг/сут) к базовым АЭП в отдельных случаях может быть эффективно при резистентных миоклонических приступах.
Бензодиазепины (клобазам 1 мг/кг/сут или клоназепам 0,5- 4,0 мг/сут) применяют у пациентов с серийными приступами и статусным течением. Назначение карбамазепина в качестве монотерапии не реко- мендовано ввиду возможной аггравации миоклонических приступов.
В лечении самого энцефалита применяются различные медикаментозные препараты: антивирусные (зидовудин, ацикловир, ганцикловир); гормональные (метилпреднизолон внутривенно 400 мг/м2 поверхности тела в течение 3 дней; преднизолон, дексаметазон); иммуноглобулины (октагам, IVIC 400 мг/кг/сут внутривенно в течение 3 дней); цитостатики (азатиоприн, циклофосфан), плазмаферез. Однако данное лечение может лишь замедлить прогрессирование заболевания.
Эффективно нейрохирургическое вмешательство - функциональная гемисферотомия, которая должна быть выполнена как можно раньше. Частота стойкой ремиссии после операции составляет 23-52%. Без оперативного лечения СР прогрессирует и заканчивается летально в течение 2-15 лет (в среднем через 3 года) с момента дебюта. Описаны отдельные случаи спонтанной стабилизации заболевания.
14.3. Идиопатические генерализованные формы эпилепсии
Доброкачественая миоклоническая эпилепсия младенчества дебюти- рует в возрасте от 4 мес до 3 лет. Характерны исключительно миоклонические приступы в виде активного миоклонуса в мышцах шеи и проксимальных отделах верхних конечностей: короткие кивки с легким наклоном туловища вперед, мгновенным приподниманием плеч и разведением локтей в стороны. Обычно приступы серийные, учащающиеся после пробуждения. Сознание не нарушено. Значительно реже наблюдаются миоклонические приступы в нижних конечностях - мгновенное сгибание ног с легким приседанием и даже возможным внезапным падением на ягодицы.
В неврологическом статусе выявляются мышечная гипотония и атаксия. Психомоторное развитие не страдает. На ЭЭГ основная активность не изменена; эпилептиформная активность регистрируется только в момент приступа. Характерны короткие разряды генерализованной полипик-волновой активности, возникающей синхронно с миоклоническими приступами. Для регистрации коротких миоклонических приступов незаменим метод видео-ЭЭГ-мониторинга. Изменения при нейровизуализации отсутствуют.
Стартовое лечение осуществляется препаратами вальпроевой кислоты. Назначают конвулекс или депакин в сиропе или каплях (после 1-2 лет - таблетированные препараты) в дозировке 300-1500 мг/сут (15-50 мг/кг/сут). В большинстве случаев наступает ремиссия. При неэффективности применяют политерапию; при этом вальпроаты всегда остаются базовыми АЭП. Назначают комбинацию вальпроатов с сукцинимидами (этосуксимид в дозе 250-750 мг/сут, 15-25 мг/кг/сут, в 2-3 приема). Возможны комбинации вальпроатов с топираматом в дозе 25-100 мг/сут (3-5 мг/кг/сут) в 2 приема; вальпроатов с бензодиазепинами, например, клобазам (фризиум) в дозе 5-20 мг/сут (0,5-1,0 мг/кг/сут) в 2 приема. Назначение карбамазепина и ламо- триджина ограничено ввиду возможности аггравации миоклонических приступов.
Прогноз благоприятный. Психическое развитие не страдает, и медикаментозная ремиссия наступает практически в 100% случаев. Продолжительность терапии - 3 года, рецидивы крайне редки.
Эпилепсия с миоклонически-астатическими приступами (синдром Доозе) дебютирует в возрастном интервале от 1 до 5 лет, чаще с генерализованных судорожных приступов, возникающих в любое время суток. В 11% случаев в анамнезе отмечаются фебрильные судороги. Типичные миоклонические и миоклонически-астатические приступы присоединяются обычно только после 3 лет. Приступы характеризуются короткими, молниеносными, обычно асинхронными и аритмичными подергиваниями в ногах и руках, чаще в проксимальных отделах. Характерно появление миоклонических «кивков», сочетающихся с легкой пропульсией туловища и при- подниманием плеч («активные кивки»). Частота миоклонических приступов может быть очень высокой; нередко приступы возникают многократно в течение одной минуты или даже постоянно, особенно после пробуждения (эпилептический статус). При миоклонических приступах в нижних конечностях возникают каскадные приседания с возможным внезапным падением на колени или ягодицы (миоклонически-астатические приступы); при этом сознание сохранено. Абсансы наблюдаются у 60-90% больных. Преобладают короткие типичные простые абсансы, а также абсансы с миоклоническим компонентом. Частота абсансов высокая, с максимумом в утренние часы.
В неврологическом статусе отмечаются односторонние пирамидные симптомы, координаторные нарушения; в половине случаев - грубая
задержка психоречевого развития. На ЭЭГ выявляются короткие генерализованные и региональные разряды пик- и полипик-волновой активности. Изменения при нейровизуализации, как правило, отсутствуют; в некоторых случаях констатируется умеренная субатрофия коры.
Стартовое лечение осуществляется препаратами вальпроевой кислоты в дозе 600-1750 мг/сут (20-100 мг/кг/сут). Препаратом второго выбора является топирамат в 2 приема в дозировках 50-200 мг/сут (3-7 мг/кг/сут). При неэффективности применяется политерапия; при этом сначала вальпроаты, а затем топирамат остаются базовыми АЭП. Применяют комбинацию вальпроатов с сукцинимидами, вальпроатов с топираматом, вальпроатов с бензодиазепинами. В отдельных резистентных случаях возможно назначение трех АЭП: вальпроатов, топирамата и сукцинимидов (или бензодиазепинов). Применение карбамазепина противопоказано ввиду возможности аггравации миоклонических приступов.
Прогноз. У большинства детей удается купировать приступы. Примерно у 1/3 пациентов эпилептические приступы сохраняются, присоединяются тонические приступы и атипичные абсансы, углубляется когнитивный дефект.
Абсансные формы эпилепсии. Наиболее частыми и хорошо изученными абсансными формами являются детская и юношеская абсансэпилепсии. Они проявляются типичными абсансами - короткими первично-генерализованными приступами с выключением сознания, замиранием, минимальными двигательными феноменами и наличием на ЭЭГ симметричной билатерально синхронной пикволновой активности с частотой 3 и более комплексов в секунду (рис. 14.2). Различают простые (замирание без двигательного компонента) и сложные (с минимальными двигательными феноменами) абсансы. К сложным относятся абсансы с тоническим (отклонение головы назад, заведение глаз вверх), миоклоническим (вздрагивание, подергивание век, бровей, крыльев носа, плеч), атоническим (падение головы на грудь, наклоны туловища), вегетативным (изменение цвета кожных покровов, непроизвольное мочеиспускание), а также с асимметричными проявлениями (например, с легким поворотом головы). Продолжительность абсансных приступов составляет от 2 до 30 с, частота - до 100 и более в сутки.
Детская абсанс-эпилепсия (пикнолепсия) - наиболее частая форма абсансной эпилепсии. Картированы мутантные гены ГАМК-рецептора в
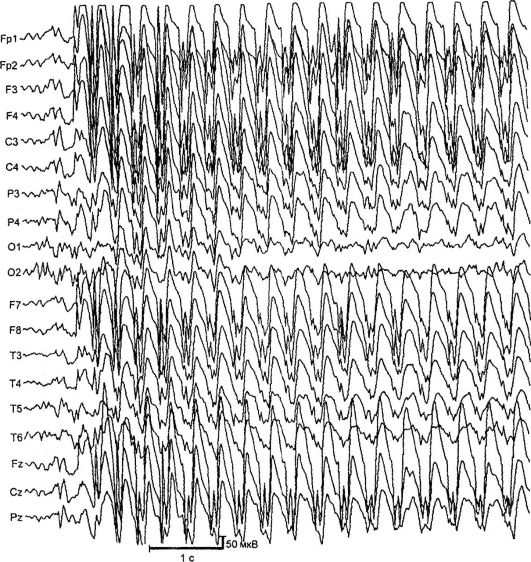
Рис. 14.2. ЭЭГ во время приступа (абсанс)
нескольких локусах хромосом: 6р, 8q24, 15q24. Заболевание дебютирует в возрасте 3-9 лет с типичных абсансов. В редких случаях заболевание начинается с генерализованных судорожных приступов с последующим присоединением абсансов. Чаще болеют девочки. Характерный тип приступов - абсансы с тоническим компонентом: легкое запрокидывание головы и заведение глазных яблок. Приступы провоцируются гипервентиляцией, реже - устным счетом. При неадекватном лечении примерно у 30% больных присоединяются ГСП. На ЭЭГ при проведении гипервентиляции появляются продолженные генерализованные разряды пик-волновой активности с частотой 3 Гц. МРТ изменений не выявляет.
Антиабсансной активностью обладают: вальпроаты, сукцинимиды, бензодиазепины, ламотриджин, топирамат. Применение препаратов
карбамазепина противопоказано, поскольку они провоцируют учащение приступов. Стартовое лечение осуществляется препаратами вальпроевой кислоты 2 раза в сутки, в дозировке 600-1800 мг/сут (30-50 мг/кг/сут). У большинства пациентов приступы полностью купируются при монотерапии вальпроатами. Препараты второго выбора - сукцинимиды. Сукцинимиды применяются в качестве монотерапии при наличии у больного изолированных абсансов, дозировка этосуксимида - 500-1000 мг/сут (15-30 мг/кг/сут) в 3 приема.
В редких резистентных случаях применяют политерапию: вальпроаты + сукцинимиды, вальпроаты и ламотриджин. Полная тера- певтическая ремиссия достигается в 90-97% случаев, обычно при монотерапии. Отмена препаратов начинается спустя 3 года после прекращения приступов.
Юношеская абсанс-эпилепсия (ЮАЭ) - форма идиопатической генерализованной формы эпилепсии, характеризующаяся типичными абсансами, дебютирующими в пубертатном периоде с высокой вероятностью присоединения ГСП и ЭЭГ-изменениями в виде коротких разрядов генерализованной быстрой пик-волновой активности. Этиология - мутация гена никотинового ацетилхолинового рецептора, связанного с хромосомами 5, 8, 18 и 21. Заболевание начинается в возрасте 9-21 года (максимум - в пубертатный период). В 40% случаев эпилепсия дебютирует с ГСП, в остальных - с абсансов. Характерны простые абсансы, меньшей продолжительности и частоты, чем при детской форме. В отдельных случаях обнаруживают очень короткие (до 3 с) абсансы с миоклоническим компонентом: замирание, легкое заведение глазных яблок вверх и быстрое подергивание век. У 75% больных наблюдается сочетание абсансов с ГСП. Судорожные приступы обычно возникают в утренние часы, после пробуждения пациентов. Частота приступов невелика - 1-4 раза в год.
ЭЭГ характеризуется нормальной основной активностью, на фоне которой выявляются короткие разряды генерализованной быстрой (4 Гц) пик-волновой активности. Большое диагностическое значение имеет появление эпилептиформной активности при депривации сна, ритмической фотостимуляции и закрывании глаз. При ЮАЭ фотосенситивность составляет 20,5%, а при ДАЭ - 10%. Проба с гипервентиляцией при ЮАЭ малоинформативна.
Стартовая терапия осуществляется препаратами вальпроевой кислоты в дозе 900-2000 мг/сут (30-40 мг/кг/сут) в 2 приема. При
отсутствии эффекта от монотерапии переходят на комбинированную терапию (вальпроаты + топирамат, вальпроаты + сукцинимид).
Полная терапевтическая ремиссия достигается в среднем у 70% больных. Отмена терапии осуществляется посте